
ABOUT US
Life is complicated interactions of various biochemical components. Recently, massively parallel sequencers have changed life science. The sequencer can quantify miscellaneous biological phenomenon. However, it is difficult to reveal biological knowledge from huge and various sequencing data for experimental biologists. In our research team, we are challenging to research and develop novel methods and software to integrate various sequencing data and understand a complex biological phenomenon at a single-cell level.
SELECTED WORK
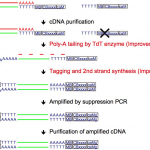 Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads.
Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads.
Sasagawa Y and Danno H. et al. developed a high-throughput single-cell RNA-seq method, Quartz-Seq2, which can analyze cells numbering up to 1,536 that are pooled together in a single sample. Quartz-Seq2 allows us to effectively utilize initial sequence reads from a next-generation sequencer. The UMI conversion efficiency in Quartz-Seq2 ranged from 32% to 35%, which is much higher than for other single-cell RNA-seq methods (7%–22%). See “Benchmarking Single-Cell RNA Sequencing Protocols for Cell Atlas Projects”.
 RamDA-seq: single-cell full-length total RNA sequencing uncovers dynamics of non-polyadenylated RNAs, recursive splicing, and enhancer RNAs.
RamDA-seq: single-cell full-length total RNA sequencing uncovers dynamics of non-polyadenylated RNAs, recursive splicing, and enhancer RNAs.
Hayashi T and Ozaki H. et al. developed Random Displacement Amplification sequencing (RamDA-seq), the first full-length total RNA sequencing method for single cells. Compared with other methods, RamDA-seq shows high sensitivity to non-poly(A) RNA and near-complete full-length transcript coverage.
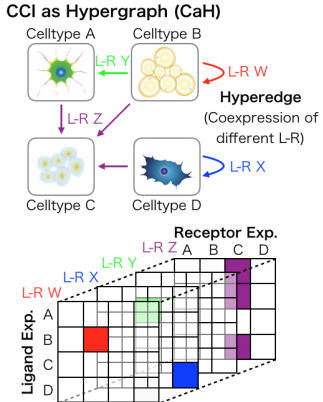 scTensor: Uncovering hypergraphs of cell-cell interaction from single cell RNA-sequencing data.
scTensor: Uncovering hypergraphs of cell-cell interaction from single cell RNA-sequencing data.
Complex biological systems can be described as a multitude of cell-cell interactions (CCIs). Recent single-cell RNA-sequencing technologies have enabled the detection of CCIs and related ligand-receptor (L-R) gene expression simultaneously. However, previous data analysis methods have focused on only one-to-one CCIs between two cell types. To also detect many-to-many CCIs, we propose scTensor, a novel method for extracting representative triadic relationships (hypergraphs), which include (i) ligand-expression, (ii) receptor-expression, and (iii) L-R pairs. When applied to simulated and empirical datasets, scTensor was able to detect some hypergraphs including paracrine/autocrine CCI patterns, which cannot be detected by previous methods.
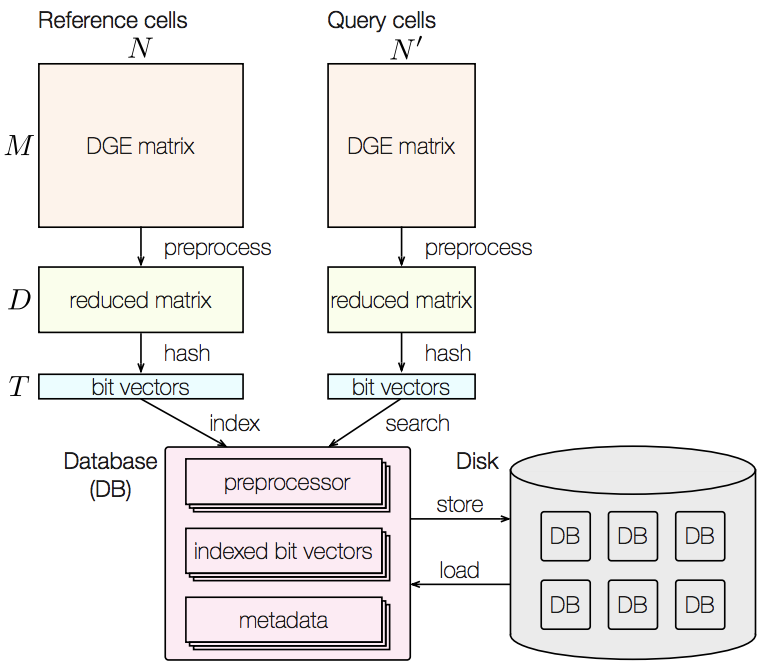 CellFishing.jl: an ultrafast and scalable cell search method for single-cell RNA-sequencing
CellFishing.jl: an ultrafast and scalable cell search method for single-cell RNA-sequencing
Recent technical improvements in single-cell RNA sequencing (scRNA-seq) have enabled massively parallel profiling of transcriptomes, thereby promiting large-scale studies encompassing a wide range of cell types of multicellular organisms. With this background, we propose CellFishing.jl, a new method for searching atlas-scale data sets for similar cells with high accuracy and throughput. Using multiple scRNA-seq data sets, we validate that our method demonstrates comparable accuracy to and is strikingly faster than the state-of-the-art software. Moreover, CellFishing.jl is scalable to more than one million cells, and the throughput of the search is approximately 1,350 cells per second (i.e., 0.74 ms per cell).
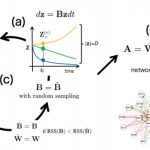 SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation.
SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation.
Matsumoto T. et. al. developed the novel and efficient algorithm SCODE to infer regulatory networks, based on ordinary differential equations. We applied SCODE to three single-cell RNA-Seq datasets and confirmed that SCODE can reconstruct observed expression dynamics. The performance of SCODE was best for two of the datasets and nearly best for the remaining dataset. We also compared the runtimes and showed that the runtimes for SCODE are significantly shorter than for alternatives.
 MeSH ORA framework: R/Bioconductor packages to support MeSH over-representation analysis.
MeSH ORA framework: R/Bioconductor packages to support MeSH over-representation analysis.
MeSH enables the extraction of broader meaning from the gene lists and is expected to become an exhaustive annotation resource for over-representation analysis (ORA). Tsuyuzaki K. et. al. developed an original MeSH ORA framework composed of six types of R/Bioconductor packages. Using our framework, users can easily conduct MeSH ORA. By utilizing the enriched MeSH terms, related PubMed documents can be retrieved and saved on local machines within this framework.
 Bayes Linux and HPC on Cloud: Bioinformatics AnalYsis Environment System on your cloud system.
Bayes Linux and HPC on Cloud: Bioinformatics AnalYsis Environment System on your cloud system.
The research project provides you a virtual machine and its chef recipes for bioinformatics analysis. The machine can work on your personal computer, private, and public cloud. Ishii M and Matsushima A. et al. provide Linux distribution and its virtual machine image with various bioinformatics tools (Debian-med and R/Bioconductor), pipeline manager (Galaxy). We also provide recipes and cookbooks to establish a data analysis environment by BioDevOps technologies.