Quartz-Seq2 is a high-throughput and high-sensitive single-cell RNA-sequencing method (using Cell sorter, UMI, Cell barcoding, and 384 well-plate).
A Workflow of Technical introduction of Quartz-Seq2
- Before the experiment, please check your experimental environments according to the guideline (page 12 in RamDA-seq protocol). To obtain a stable result, you should always store all enzymes at -30ºC using a cold tube rack, such as NEB cooler (T7771) or -20ºC labtop cooler.
- First, you should check that the Quartz-Seq2-WTA (whole-transcript amplification) reaction works well using the operation check protocol. If you succeed in amplifying, you will obtain 25-50ng WTA cDNA per technical replicate. This result means that your prepared reagent, primer, pieces of equipment, etc., work well.
- After that, you should analyze10 pg of total RNA in all 384-wells at approximately 0.1 M fastq reads on average per well using 384_indexes protocols. You will obtain 50-70ng WTA cDNA per 384-well plate. Please confirm that UMI conversion efficiency levels of Quartz-Seq2 are over 30%.
Operation Check Protocol
Quartz-Seq2 Protocol (384 well plate)
English
Japanese
- 2.0.2_Quartz-Seq2_384indexes_protocol_Jp (Current)
- 2.0.1_Quartz-Seq2_384indexes_protocol_Jp
- 2.0.0_Quartz-Seq2_384indexes_protocol_Jp
Video Tutorial
A cryopreservation step of 384 multiwell plates just after single-cell sorting.
How to assemble collector unit for pooling of cell-barcoded cDNA.
Software
Data analysis workflow for Quartz-Seq2 (GitHub)
Data
- SRA files and gene expression matrices of Quartz-Seq2 (NCBI GEO)
- FASTQ files (ENA)
- Class labeled digital expression matrix
- Allow list (List of cell barcodes)
Accessories
- Metal frame for spin-down collection system (Fig. S4A)
- Cooling adopter for Formulatrix MANTIS® Liquid Handler
FAQ
Q1. What do you mean “distance of SeqLv was 5”?
A1. SeqLv means Sequence Levenshtein distance. “edit distance 5” indicates that two deletions, insertions, or mismatch of cell barcode can be informatically corrected. There is a similar distance, the so-called Humming distance to SeqLv. However, Humming distance can not correct two deletions or insertions in cell barcode sequences.
Q2. Could you clarify the composition of the TdT buffers RH55 and T55 in your Quartz-Seq2 paper?
A2. Please see as follows. T100 is available as 1xThermopol Buffer from NEB company. RH100 is available as 1xRNase H buffer from NEB company.
| T100 | 20mM Tris-HCl (pH8.8), 10mM (NH4)2SO4, 10mM KCl, 2mM MgSO4, 0.1% Triton X-100 |
| T55 | 11mM Tris-HCl (pH8.8), 5.5mM (NH4)2SO4, 5.5mM KCl, 1.1mM MgSO4, 0.055% Triton X-100 |
| RH100 | 50mM Tris-HCl (pH 8.3), 75mM KCl, 3mM MgCl2, 10mM DTT |
| RH55 | 27.5mM Tris-HCl (pH 8.3), 41.25mM KCl, 1.65mM MgCl2, 5.5mM DTT |
- https://www.neb.com/products/b9004-thermopol-reaction-buffer#Product%20Information
- https://www.neb.com/products/m0297-rnase-h#Product%20Information
Q3. Where can I buy a metal frame for spin-down collection system? (Fig. S4A)
A3. Please contact
Operation check:
3 times- Swing, 4ºC, 1500g, 10min (Metal frame + One well reservoir + 384 well PCR plate) *
3 times- Swing, 4ºC, 2000g, 10min (Metal frame + One well reservoir + 384 well PCR plate)
3 times- Swing, 4ºC, 2500g, 10min (Metal frame + One well reservoir + 384 well PCR plate)
3 times- Swing, 4ºC, 3000g, 10min (Metal frame + One well reservoir + 384 well PCR plate)
*: Recommended
The step of the spin-down collection can be completed by 1,500g at 4ºC> for 3-5min.
Q4. Could you show the list of reagents for Quartz-Seq2?
A4. Please see the following file. Additional file 4: Table S3.
Q5. Can I change from X reagent to Y reagent in Quartz-Seq2?
A5. No. It is no longer Quartz-Seq2. We cannot support your original scRNA-seq method 🙂
Q6. Which should I use 384 multiwell plates?
A6. We strongly recommend twin.tec PCR Plate 384 clear (0030 128.508, Eppendorf).
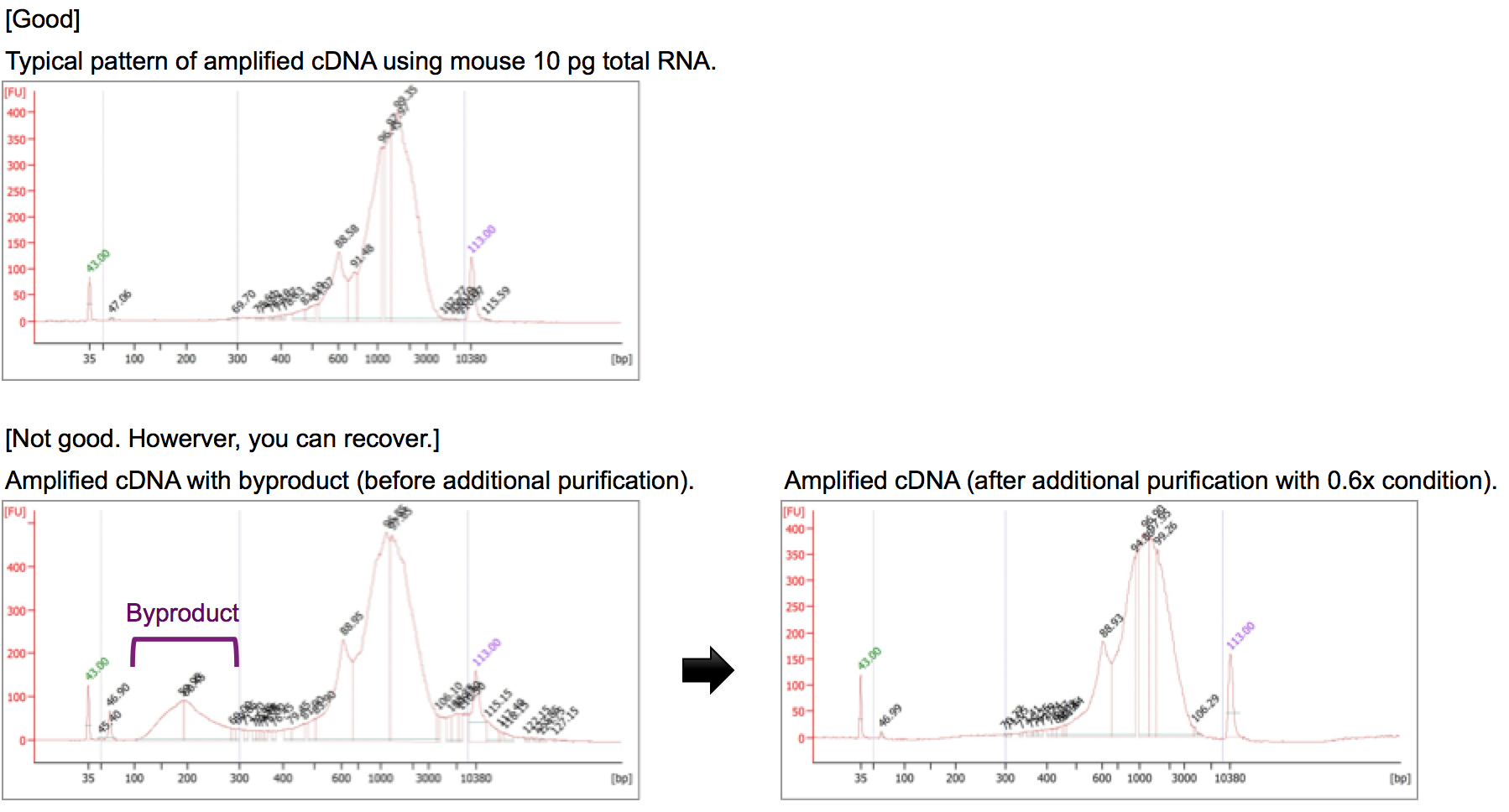
Q7. What is the byproduct of whole-transcript amplification in Quartz-Seq2? Can I remove the byproduct from amplified cDNA?
A7. It has been reported that byproducts could be synthesized from reverse-transcription (RT) primers in poly-A tagging-based methods, including Quartz-Seq2. Almost RT primers can be removed at the cDNA purification step. Therefore, there is no byproduct in amplified cDNA, typically. If you observe the byproduct, please remove the byproduct by additional purification with 0.6-0.7x Ampure XP beads.
Q8. How do we correct the cell barcode in Quartz-Seq2?
Please use correct_barcode. If you should de-multiplex a fastq file from Quartz-Seq2, please use demultiplexer_quartz-seq2. We don’t recommend to demultiplex a fastq file of Quartz-Seq2. Typical pipelines for high throughput scRNA-seq, including Drop-seq pipeline or CellRenger, require a fastq file before demultiplexing.
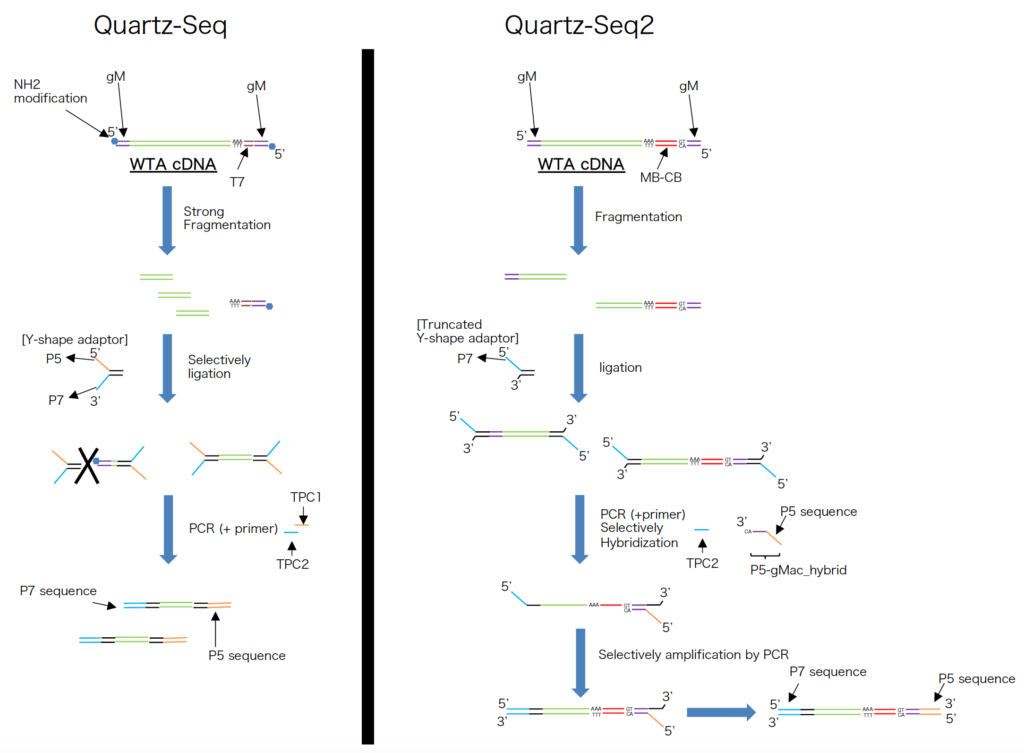
Q9. Show us the schematic representation of sequence library preparation for Quartz-Seq and Quartz-Seq2.
Please see the below figure.
Citation
- Yohei Sasagawa, Hiroki Danno, Hitomi Takada, Masashi Ebisawa, Kaori Tanaka, Tetsutaro Hayashi, Akira Kurisaki, Itoshi Nikaido. Quartz-Seq2: a high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads. Genome Biology 2018 19:29
- Yohei Sasagawa, Tetsutaro Hayashi and Itoshi Nikaido. Strategies for converting RNA to amplifiable cDNA for single-cell RNA sequencing methods. Single Molecular and Single Cell Sequencing. Advances in Experimental Medicine and Biology. Springer (Invited review). Apr 10 2019.
- Elisabetta Mereu, Atefeh Lafzi, Catia Moutinho, Christoph Ziegenhain, Davis J. McCarthy, Adrian Alvarez, Eduard Batlle, Sagar, Dominic Grün, Julia K. Lau, Stéphane C. Boutet, Chad Sanada, Aik Ooi, Robert C. Jones, Kelly Kaihara, Chris Brampton, Yasha Talaga, Yohei Sasagawa, Kaori Tanaka, Tetsutaro Hayashi, Caroline Braeuning, Cornelius Fischer, Sascha Sauer, Timo Trefzer, Christian Conrad, Xian Adiconis, Lan T. Nguyen, Aviv Regev, Joshua Z. Levin, Swati Parekh, Aleksandar Janjic, Lucas E. Wange, Johannes W. Bagnoli, Wolfgang Enard, Marta Gut, Rickard Sandberg, Itoshi Nikaido, Ivo Gut, Oliver Stegle, Holger Heyn. Benchmarking Single-Cell RNA Sequencing Protocols for Cell Atlas Projects. Nature Biotechnology. 06 April 2020.
Logs for Quartz-Seq2
The following are the logos of Quartz-Seq1/2. You are free to use them as long as you do not change the design.











Media
- RIKEN press release (Japanese). 2018/03/13.
- RNA-seq blog. 2018/03/13.
- RIKEN press release. 2018/5/15.
If you need to detect full-length total RNA at a single-cell level, please see the RamDA-seq page.
Contact
support-bit (at) riken (dot) jp
If you are a commercial company member and are interested in collaborating with us on Quartz-Seq2, please contact Knowledge Palette, Inc.