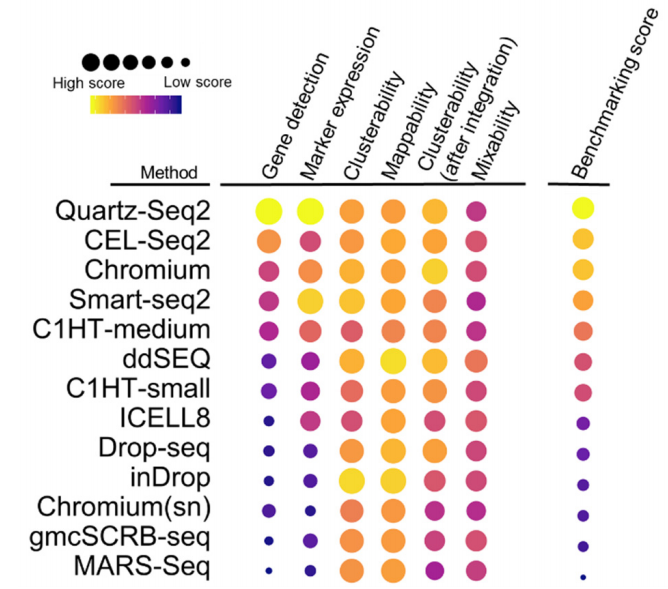
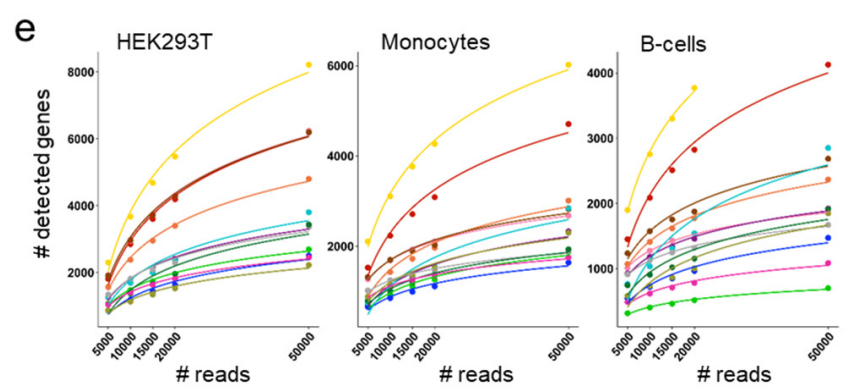
Elisabetta Mereu, Atefeh Lafzi, Catia Moutinho, Christoph Ziegenhain, Davis J.MacCarthy, Adrian Alvarez, Eduard Batlle, Sagar, Dominic Grün, Julia K. Lau, StéphaneBoutet, Chad Sanada, Aik Ooi, Robert C. Jones, Kelly Kaihara, Chris Brampton, YashaTalaga, Yohei Sasagawa, Kaori Tanaka, Tetsutaro Hayashi, Itoshi Nikaido, CorneliusFischer, Sascha Sauer, Timo Trefzer, Christian Conrad, Xian Adiconis, Lan T. Nguyen, Aviv Regev, Joshua Z. Levin, Aleksandar Janjic, Lucas E. Wange, Johannes W. Bagnoli, Swati Parekh, Wolfgang Enard, Marta Gut, Rickard Sandberg, Ivo Gut, Oliver Stegle, Holger Heyn. Benchmarking Single-Cell RNA Sequencing Protocols for Cell Atlas Projects. bioRxiv. (submitted)
生命科学のデータは、ゲノム、RNA、タンパク質、表現型など様々な階層に渡り、複雑で多様なデータ構造を扱います。生命科学分野でのデータ解析では、これらのデータを統合的に解析する手法の確立が大きな課題となっています。一方、機械学習の分野では、Heterogeneous Information Network (HIN) という分野で、このような複雑なデータを統合的に解析する手法が発達しつつあります。