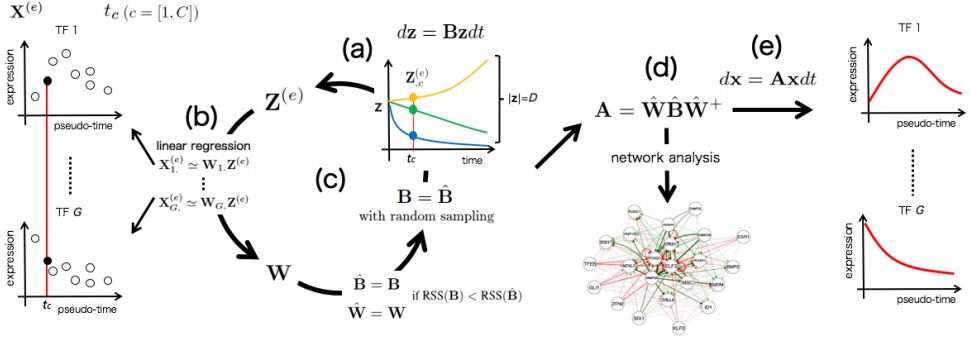
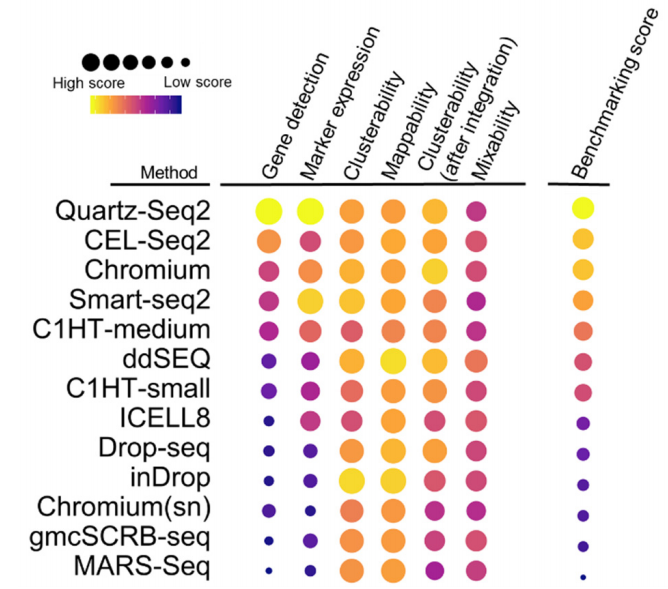
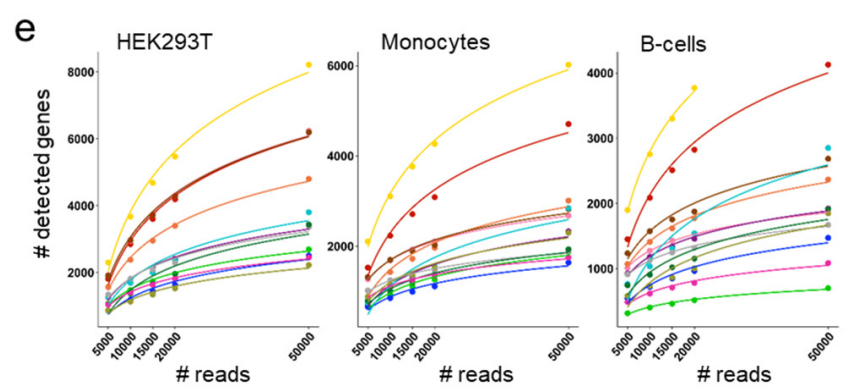
Elisabetta Mereu, Atefeh Lafzi, Catia Moutinho, Christoph Ziegenhain, Davis J.MacCarthy, Adrian Alvarez, Eduard Batlle, Sagar, Dominic Grün, Julia K. Lau, StéphaneBoutet, Chad Sanada, Aik Ooi, Robert C. Jones, Kelly Kaihara, Chris Brampton, YashaTalaga, Yohei Sasagawa, Kaori Tanaka, Tetsutaro Hayashi, Itoshi Nikaido, CorneliusFischer, Sascha Sauer, Timo Trefzer, Christian Conrad, Xian Adiconis, Lan T. Nguyen, Aviv Regev, Joshua Z. Levin, Aleksandar Janjic, Lucas E. Wange, Johannes W. Bagnoli, Swati Parekh, Wolfgang Enard, Marta Gut, Rickard Sandberg, Ivo Gut, Oliver Stegle, Holger Heyn. Benchmarking Single-Cell RNA Sequencing Protocols for Cell Atlas Projects. bioRxiv. (submitted)
大野圭一朗
University of California, San Diego Trey Ideker Lab
/ National Resource for Network Biology / The Cytoscape Consortium 「バイオインフォマティクス分野における可視化アプリケーション構築と維持の実際」